真核细胞延伸因子2激酶(eEF2K)是由钙/钙调素激活的α-激酶家族的成员。作为翻译的关键调节因子,eEF2K 可使 eEF2 在其成熟多肽的 Thr56 氨基酸残基上磷酸化,从而降低其对核糖体的亲和力,抑制蛋白质合成的延长。值得注意的是,eEF2K在恶性细胞中经常高度过表达。eEF2K 对肿瘤细胞的许多细胞过程至关重要,包括调节自噬活性和细胞 ATP 水平、血管生成、肿瘤细胞迁移、增殖、存活和上皮-间质转化(EMT)。因此,eEF2K 被认为是抗癌治疗的有效靶点。
自 1994 年发现首个强效 eEF2K 抑制剂 Rottlerin 以来,在过去二十年中,有关 eEF2K 抑制剂的报道越来越多。目前已发现多种小分子化合物可通过靶向和抑制 eEF2K 的活性来影响癌症的进展。如高选择性单靶点 eEF2K 抑制剂 A-484954、被广泛接受的哺乳动物 eEF2K 抑制剂 NH125、以及双靶点抑制剂 18i。一些新兴的抑制策略,如 PROTAC 策略和联合治疗药物设计,为未来的癌症治疗带来了巨大希望。总之,这些鼓舞人心的发现将为用小分子化合物抑制 eEF2K 的有前景的策略提供新的见解,以大大改善未来的癌症治疗。
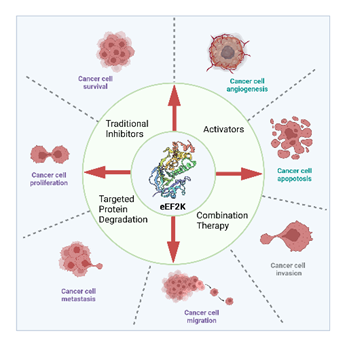
图1 eEF2K与癌症进程间的关系及其靶向小分子抑制剂开发中的主要类型
近日,9778818威尼斯符雷蕾团队在美国药学权威期刊Drug Discovery Today上发表了一篇题为“Targeting eukaryotic elongation factor 2 kinase (eEF2K) with small-molecule inhibitors for cancer therapy”综述(https://doi.org/10.1016/j.drudis.2024.104155. 影响因子:6.5)。这篇综述总结了eEF2K 的分子结构及其在癌症中的致癌作用,包括血管生成、肿瘤细胞迁移、增殖、存活和上皮-间质转化(EMT),同时进一步讨论了针对eEF2K的小分子化合物的现状,包括单靶点、双靶点小分子化合物以及新出现的联合治疗策略,以期为肿瘤治疗提供新思路。论文第一作者为9778818威尼斯2023级硕士研究王会苹,最后通讯作者为符雷蕾,9778818威尼斯为第一署名单位和通讯作者单位。

一、 eEF2K的分子结构和调节位点
现有研究结果表明,eEF2K的催化结构域与传统真核生物蛋白激酶的催化结构域没有同源性。其特殊的催化结构域能识别底物α螺旋内的氨基酸并使之磷酸化,因此被定位为一个新的非典型α激酶超家族的成员。eEF2K由一个位于N端的非典型CaM 结合域(CTM)组成,通过一个调节元件(RE)和一个与 C端域(CTD)相连的长调节环(R-loop)与α激酶域(KD)相连。据预测,CTD 包含三个螺旋重复,编码底物eEF2的结合位点。eEF2K的催化结构域位于分子N端的 77-99个残基,CaM与eEF2K催化结构域的末端结合、eEF2K的残基 562-725包含两个 α-helical 重复序列,具有TPR 结构域及其 SEL1-ike 亚家族的特征。eEF2K的 SEL1 样结构域是底物磷酸化所必需的。N/D 环中高度保守的残基对酶的活性至关重要,R环包含多个磷酸化位点。
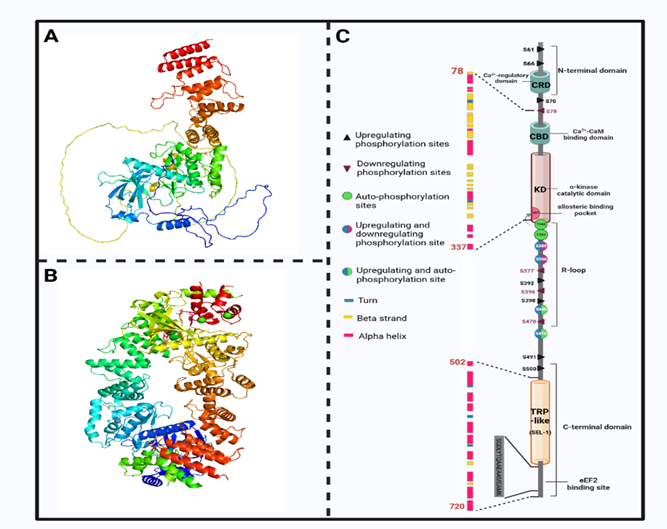
图2 eEF2K分子结构及调节位点
二、 eEF2K在癌症中的关键作用途径
作者也对eEF2K在癌症中的关键作用途径进行了总结。高度调控的蛋白激酶 eEF2K几乎完全依赖于 Ca2+/钙调蛋白(CaM)。在 Ca2+ /CaM存在的情况下,eEF2K在多个位点进行自身磷酸化,以获得最大活性。Akt/GSK3/eEF2K 信号通路通过磷酸化 Ser392 来调节 eEF2K。AMPK还能直接磷酸化 eEF2,通过多位点磷酸化,AMPK 触发 eEF2K 激活。在体外,eEF2K被 AMPK在 Ser398 处磷酸化,这被认为是 eEF2K 激活的原因。此外,eEF2K还是MAPK和PI3K通路下游的一个关键汇合点。作为 PKC 的下游蛋白被激活。mTORC1由生长因子、激素或氨基酸激活,是细胞营养或能量状况的重要传感器。eEF2K 位于 mTOR 的下游,mTORC1激活后,eEF2K在 Ser70、Ser78、Ser359、Ser392、Ser396 和 Ser470 处磷酸化,导致 eEF2K 失活,并阻止 eEF2 的磷酸化,从而导致 eEF2 水平升高并诱导蛋白质合成。FOXM1 是第一个驱动eEF2K 表达的转录因子。eEF2K mRNA 的启动子包含几个结合位点,FOXM1与这些位点直接结合,转录调控eEF2K基因的表达。NF-κB的激活会抑制 eEF2K 的转录,导致 eEF2K 蛋白水平下降。
三、 eEF2K的致癌作用
此外,作者完整地总结了eEF2K 在癌症发展过程中的促进作用机制。eEF2K可增加细胞增殖和肿瘤生长,快速增殖的恶性细胞表现出eEF2K 活性水平升高。它以CaM 依赖性方式激活ERK、Akt、p38和HSP27信号(图2C)。通过阻断细胞周期蛋白 D1、Src 和 MAPK/ERK信号转导,eEF2K 的表达可调节肺癌细胞的增殖、侵袭和肿瘤发生。通过eEF2K/p-STAT3 通路,eEF2K可促进沃伯格效应,抑制肺癌细胞的有糖酵解,从而阻碍细胞和肿瘤的生长和增殖。癌细胞通过 eEF2K/TG2轴诱导凋亡诱导因子,导致不依赖于 caspase 的细胞凋亡。eEF2K通过 eEF2K/ATG7 调节参与自噬体结构形成的关键自噬基因的表达。eEF2K 同时调节内在途径和外在途径,以刺激细胞凋亡。
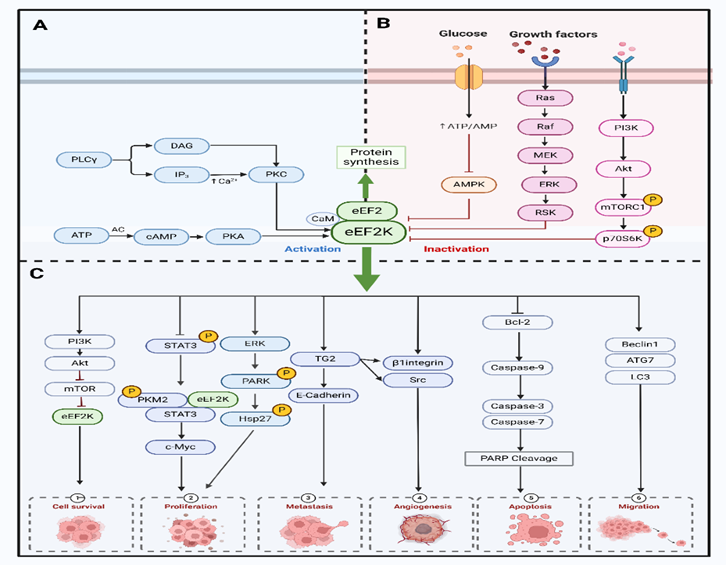
图3 eEF2K在癌症中的关键作用途径
四、 针对 eEF2K 的小分子抑制剂用于癌症治疗
在该综述中,作者系统地总结了eEF2K 的小分子抑制剂用于癌症治疗的现状。作为一种非典型激酶,eEF2K与其他蛋白激酶不同,因此能够开发出具有强效选择性而不影响其他激酶的小分子抑制剂。然而,eEF2K不受"经典"蛋白激酶抑制剂的抑制。此外,由于缺乏eEF2K及其催化结构域的三维结构信息,开发小分子抑制剂的工作变得更具挑战性。虽然已经合成了几种针对eEF2K的抑制剂,但没有一种具有特异性或足够有效。以eEF2K 为靶点的传统小分子化合物传统的小分子化合物通常是eEF2K单靶点抑制剂。许多天然或合成化合物已被用于靶向 eEF2K。这些化合物通过不同的机制抑制 eEF2K,如干扰 CaM 结合位点、ATP 结合位点或 eEF2 结合位点。
更重要的是,随着包括 PROTAC 和基因编辑在内的越来越多的新技术被应用于药物开发,人们有机会发现更具特异性和效力的eEF2K小分子抑制剂,作为未来的癌症治疗药物。由于肿瘤发生过程受到多种因素的影响,只针对一种因素很可能无法遏制癌症的发展。因此,针对一种以上的癌症相关因素进行综合治疗似乎可以达到控制癌症的目的。
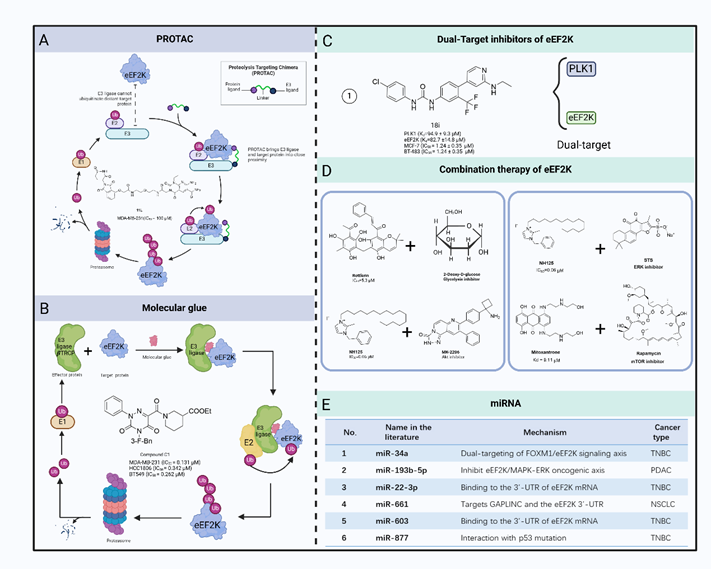
五、 结论与展望
目前,人们对 eEF2K 抑制剂治疗癌症的有效性还存在争议。此外,eEF2K的完整三维结构仍未解决,因此制约了通过基于结构的药物设计开发eEF2K抑制剂。目前对新型eEF2K抑制剂既能有效抑制 eEF2K,又具有低细胞毒性或无细胞毒性的需求尚未得到满足。仍迫切需要将针对各种癌症的eEF2K靶向药物进行临床转化。幸运的是,越来越多的新技术包括用于药物开发的 PROTAC、基因编辑和非编码RNA。与传统的药物开发技术相比,新药已显示出良好的抑制效果和令人信服的安全性和耐受性。基因编辑技术通过准确定位和修改突变细胞中的异常基因,实现直接、持续的干预。与RNA干扰蛋白质降解技术相比,靶向蛋白质解释技术具有降解速度更快、特异性更强、可逆性更好等优势。
原文链接:https://www.sciencedirect.com/science/article/pii/S1359644624002800
{kind=link}
